

Śródbłonek naczyniowy
Śródbłonek naczyniowy spełnia wiele kluczowych funkcji [1,3] dla utrzymania homeostazy naczyń m.in. bierze udział w przekazywaniu bodźców zarówno humoralnych, jak i mechanicznych, poza tym odpowiada także m.in. za syntezę tlenku azotu (NO) oraz śródbłonkowego czynnika wzrostu fibroblastów (PDGF). Zaburzenie funkcji śródbłonka jest czynnikiem patogennym leżącym u podłoża wielu chorób naczyniowych, w tym także miażdżycy naczyń.
W przypadku miażdżycy istnieją w tętnicach miejsca predysponujące do rozwoju jak rozgałęzienia czy kręty przebieg naczyń, w których dochodzi do zaburzeń przepływy krwi oraz sił ścinających, predysponując do przenikania lipopolisacharydów o niskiej gęstości (LDL) zawierających apolipoproteinę B (apoB).
W ostatnich latach w badaniach [1,3] na ludzkich tętnicach szyjnych oraz aortach myszy zaobserwowano istnienie miejsc opornych oraz wrażliwych na powstawanie zmian miażdżycowych. Obserwacje te są związane z budową strukturalną, molekularną oraz funkcjonalną komórek śródbłonka.
W miejscach opornych uwidoczniono komórki śródbłonka o budowie elipsoidalnej ułożone zgodnie z kierunkiem przepływu krwi, natomiast w regionach podatnych na miażdżycę występowały komórki o budowie sześciennej.
W innych badaniach zaobserwowano, że komórki śródbłonka pokryte nienaruszoną warstwą glikokaliksu wykazują większą odporność na przenikanie LDL pod śródbłonek.
Przyglądając się bliżej pod kątem molekularnym dostrzeżono różnice w ekspresji genów pomiędzy miejscami opornymi i wrażliwymi na powstawanie blaszki miażdżycowej.
Najbardziej widoczna jest aktywacja ekspresji czynników transkrypcyjnych KLF2 oraz KLF4 (Kruppel-like factor 2 i 4) jako działanie ochronne w miejscach opornych oraz aktywacja szlaku NF-κB jako czynnika promującego powstawanie miażdżycy. Ekspresja KLF2 i KLF4 aktywuje szlaki sygnalizacyjne MEK5, ERK5 oraz MEF2, ponadto może być modulowana poprzez kinazę AMP, SIRT1, kinazę białkową C, proteazę specyficzną SUMO 2, deacetylazę histonów 5, oraz mikro-RNA m.in. miR-92, miR-126 [6].
Ekspresja KLF2 jest widoczna także w innych komórkach związanych z miażdżycą m.in. monocytach, limfocytach T i komórkach dendrytycznych wykazując działanie przeciwzapalne. Poza tym warto zauważyć, że zgodnie z wynikami badań, jednym z plejotropowego działania statyn – grupy leków stosowanych w celu obniżenia endogennej syntezy cholesterolu – jest wzrost ekspresji genu dla KLF2 [3].
W przeciwieństwie do tego czynnik NF-κB bierze udział już we wczesnych etapach tworzenia blaszki miażdżycowej poprzez aktywację ekspresji pro-miażdżycowych receptorów takich jak VCAM-1 czy TLR2. NF-κB, wzmaga także produkcję w śródbłonku cytokin i chemokin prozapalnych, białek macierzy zewnątrzkomórkowej, czynników wzrostu oraz mikro-RNA [6]. Funkcję regulacyjną pełni tutaj podobnie jak poprzednio mikro-RNA np. miR-181b czy 146a [6]. Natomiast za regulację potencjału redoks w komórce i dzięki temu działanie miażdżyco-protekcyjne oraz chroniące przed stresem oksydacyjnym odpowiada czynnik jądrowy 2 (Nrf2) [3]. Zaobserwowano jego ekspresję w miejscach opornych na rozwój choroby, a także wykazano, że do pełnego działania antyoksydacyjnego wymaga ekspresji wspomnianego czynnika KLF2.
Ostatnio zaobserwowano, że zmiana charakteru przepływu pobudza białko TIAM1, które następnie pobudza PECAM-1 tworzące kompleks z VE-kadheryną oraz VEGFR2 w komórkach śródbłonka. W kolejnym etapie aktywacja małej GTPazy Rac1 przez PECAM-1 powoduje aktywację szlaku NF-κB oraz produkcji reaktywnych form tlenu.

Poza tym wykazano związek pomiędzy działaniem sił ścinających w naczyniach a zmianą metylacji DNA – głównie poprzez aktywację DNMT1 – zwiększając w ten sposób metylację regionu bliższego promotora dla KLF4 i hamując jego ekspresję. Zaburzenia przepływu mogą również wpływać na splicing mRNA hamując ochronny splicing pomiędzy egzonami EIIIA i EIIIB dla genu fibronektyny.
Istnieje również korelacja pomiędzy aktywacją białka G sprzężonego z S1P1 w śródbłonku naczyń a regulacją przepływu krwi. Posługując się modelem zwierzęcym, stosując knockout genu dla S1P1 w naczyniach siatkówki u myszy zaobserwowano zwiększenie fosforylacji endotelialnej syntazy tlenku azotu (eNOS), co skutkowało obniżeniem syntezy NO.
Udowodniono również udział kanału jonowego aktywowanego mechanicznie PIEZO1 w regulacji aktywności kalpainy i regulacji przepływu krwi poprzez wpływ na napływ jonów wapnia do komórek śródbłonka.
Kolejnym czynnikiem jest Syndekan 4 – przezbłonowy proteoglikan siarczanu heparanu – w badaniach na myszach pozbawionych ekspresji genu dla Syndekanu 4, zaobserwowano wzrost powstawania zmian miażdżycowych w porównaniu do kontroli.
W ostatnio opublikowanych badaniach [5] wykazano ciekawą zależność pomiędzy kinazą MAP4K4 – należącą do rodziny kinaz aktywowanych mitogenami (MAP), a progresją zmian miażdżycowych. Na podstawie badań na modelu mysim oraz ludzkich komórkach, wykazano, że zastosowanie selektywnego inhibitora MAP4K4 powoduje regresję zmian miażdżycowych. Na chwilę wydaje się, że będzie to ważny punkt uchwytu nowych leków zmniejszających rozwój miażdżycy.
Makrofagi
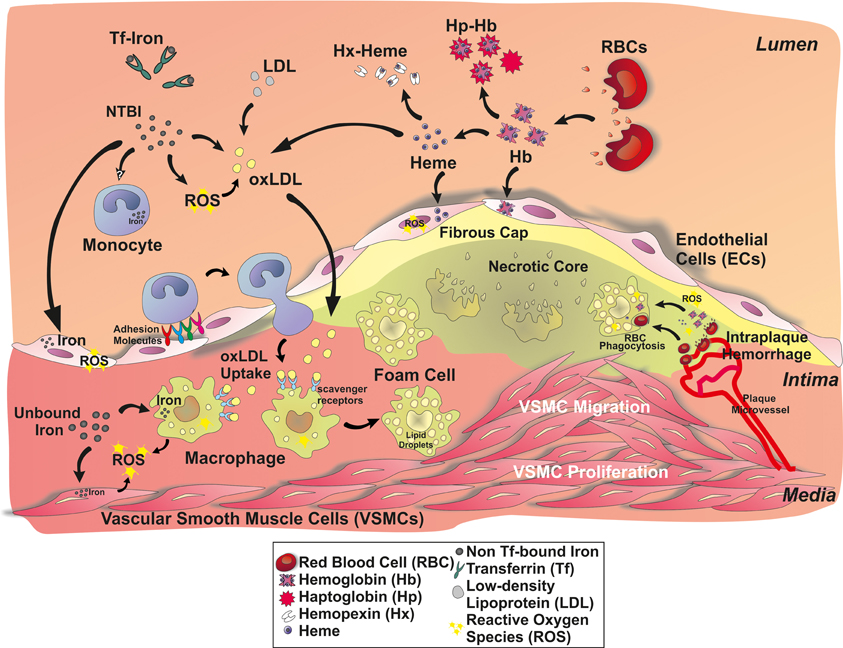
Udział makrofagów jest kolejnym etapem progresji miażdżycy, biorącym udział w powstawaniu blaszki miażdżycowej [1,4]. W odpowiedzi na chemokiny wydzielane w śródbłonku naczyń pod wpływem kumulujących się podśródbłonkowo LDL, następuje stymulacja komórek macierzystych szpiku kostnego i uwalnianie monocytów. Następnie dojrzewają i przekształcają się w makrofagi zdolne do fagocytowania skumulowanych podśródbłonkowo LDL zarówno natywnych, jak i poddanych utlenianiu (oxLDL) oraz innym modyfikacjom. W badaniach postulowany jest udział zarówno fagocytozy za pośrednictwem receptorów typu scavenger, jak i pinocytozy. W makrofagach pochłonięte LDL zostają skierowane do późnych endosomów oraz lizosomów, gdzie są przekształcane przez hydrolazy lizosomalne. Niestrawiony cholesterol jest estryfikowany i magazynowany w komórce w postaci kropelek lipidowych przypominających pianę – tworząc komórki piankowate. Co ciekawe w hodowli komórkowej zaobserwowano, że hydroliza może także zachodzić pozakomórkowo w odgrodzonych, kwaśnych przedziałach.
Makrofagi kumulujące się w blaszkach miażdżycowych, na drodze aktywacji szlaków zapalnych, wpływają na śródbłonek powodując dalszy napływ monocytów/makrofagów.
Zaobserwowano także, że mitochondrialny stres oksydacyjny w hodowlach makrofagów może być indukowany poprzez oxLDL oraz sterole, aby dzięki aktywacji szlaku NF-κB stymulować produkcję chemokiny MCP-1 i dalszy napływ monocytów.

Końcowy efekt promiażdżycowy aktywowanych makrofagów jest zależny od równowagi pomiędzy działaniem prozapalnym oraz hamującym odpowiedź zapalną. Makrofagi zawierające oxLDL, utlenione fosfolipidy czy z nadmierną akumulacją cholesterolu m.in. poprzez uszkodzenie transporterów z rodziny kaset wiążących ATP (ABCA1 oraz ABCG1) - odpowiadających za transport nadmiaru cholesterolu z komórek do lipoprotein o wysokiej gęstości (HDL) - ulegają apoptozie i są fagocytowane przez inne makrofagi na drodze efferocytozy. Następnie akumulacja cholesterolu w komórce powoduje zahamowanie jego endogennej syntezy głównie poprzez wpływ na enzym reduktazę 24-dehydrocholesterolu i nagromadzenie produktu pośredniego – desmosterolu, który wykazuje działanie przeciwzapalne - dzięki oddziaływaniu na wątrobowy X receptor (LXR) pośredniczący w odpowiedzi przeciwzapalnej.
W chorobowo zmienionych makrofagach przez nagromadzenie zestryfikowanego cholesterolu białko homologiczne C/EBP (CHOP) związane ze stresem w retikulum endoplazmatycznym (ER) aktywuje receptor dla 3-fosfatydylo inozytolu otwierający kanał wapniowy w ER, powodując uwalnianie wapnia do cytoplazmy. Zwiększone stężenie jonów wapnia aktywuje proteinazę II zależną od wapnia i kalmoduliny (CaMKII), wpływając na indukcję szlaku mitochondrialnego apoptozy. Jednocześnie CHOP wpływa na zmniejszenie ekspresji białka antyapoptotycznego Bcl-2. Następuje także aktywacja TLR4 oraz dysfunkcja lizosomów, która jest kluczowa dla przekształcania cholesterolu i powoduje dalsze nagromadzenie zestryfikowanego cholesterolu w postaci kropel lipidowych.
Istnieją także dowody na aktywację inflamasomu w zmianach miażdżycowych i prawdopodobnie IL-1β ma także swój udział w powstawaniu miażdżycy. Obserwacje in vitro oraz in vivo pozwalają na postawienie hipotezy, że prawdopodobnie kryształki cholesterolu, które nie uległy strawieniu w makrofagach kierują komórkę w stronę aktywacji inflamasomu.
Poza tym zarówno aktywacja CD36 oraz utlenione cząsteczki DNA mitochondrialnego mogą aktywować inflamasom.
W przypadku uszkodzenia mechanizmu efferocytozy, np. w efekcie niedotlenienia zmian miażdżycowych, makrofagi umierają na drodze nekrozy promując sygnalizację prozapalną wzorców molekularnych związanych z uszkodzeniem (DAMP).
Makrofagi są źródłem prozapalnych cytokin i chemokin jak np. IL-1, IL-6, IL-12, IL-15, IL-18, TNF-α, ligandu chemokin 2 (CCL2). Uwidoczniono również działanie promiażdżycowe receptora chemokin CXCR2 – na modelu mysim z deficytem CXCR2 zauważono zmniejszone zmiany miażdżycowe oraz zmniejszoną akumulację makrofagów. Ostatnio zaobserwowano, że IL-17A promuje u myszy adhezję monocytów, a także po związaniu ze swoim receptorem powoduje w końcowej reakcji aktywację czynnika NF-κB. IL-23 natomiast prawdopodobnie promuje powstawanie zmian martwiczych poprzez pośredniczenie w apoptozie makrofagów.
Poza czynnikami prozapalnymi, wydzielają także mediatory przeciwzapalne jak np. IL-10, IL-13, IL-27, TGF-β oraz CXCL5, który odpowiada za indukcję ABCA1 w makrofagach.
Ustala się także równowaga pomiędzy syntezą w makrofagach prozapalnego leukotrienu B4 oraz przeciwzapalnego lipoksyny A4 przy udziale 5-lipooxygenazy.
Makrofagi mają również ścisły związek z ostrymi zdarzeniami miażdżycowo-zakrzepowymi - przyczyniają się do ścieńczenia włóknistej czapeczki zabezpieczającej zmianę miażdżycową przed pęknięciem i przedostaniem się do światła naczynia poprzez wydzielanie metaloproteinaz macierzy (MMP).

Mięśnie gładkie naczyń
Stabilność blaszki miażdżycowej w dużej mierze zależy od grubości ochronnej włóknistej czapeczki oraz stopnia zapalenia. Pęknięcia poza działaniem MMP są spowodowane zmniejszeniem grubości wspomnianej warstwy ochronnej spowodowanej przez śmierć komórek mięśni gładkich naczyń (VSMC), rozpad kolagenu oraz macierzy zewnątrzkomórkowej. W związku z tym wydaje się, że komórki VSMC pełnią funkcję ochronną przed zdarzeniami zakrzepowo-zatorowymi dzięki stabilizacji blaszki miażdżycowej – współdziałając w tworzeniu warstwy ochronnej.
Jak zaobserwowano [1,2], VSMC pochodzą z różnych multipotencjalnych prekursorów, w zależności od miejsca np. wieńcowe VSMC pochodzą z nasierdzia, a VSMC aorty zstępującej głównie z prekursorów somatycznych.
Fenotypowo wszystkie populacje VSMC wyglądają podobnie, jednak mogą występować różnice w odpowiedzi na czynniki regulacyjne. Jednym z przykładów jest zaobserwowana u myszy z wyciszonym genem dla apolipoproteiny E (apoE) poddanych diecie wysoko-tłuszczowej większa ekspresja genów Homeobox (Hox) w miażdżyco-opornej aorcie piersiowej, w stosunku do podatnego na miażdżycę łuku aorty oraz odwrotną zależność w przypadku czynnika NF-κB. Podobne różnice obserwowano in vitro w modelu ludzkich komórek VSMC.
W badaniach dotyczących roli VSMC w patogenezie miażdżycy, występują pewne trudności ze względu na zdolność ekspresji markerów zarówno dla mięśni gładkich, jak i dla makrofagów, przez co nie można ze stuprocentową pewnością określić, z którego typu pierwotnie wywodzą się badane komórki. W ostatnich badaniach komórek piankowatych z obszaru zaawansowanych zmian miażdżycowych wykazały ekspresję zarówno markera ACTA2 charakterystycznego dla SMC, jak i CD68 będącego markerem makrofagów. W innych badaniach natomiast zaobserwowano dodatkowo ekspresję markerów epigenetycznych charakterystycznych dla komórek mięśni gładkich – MYH11 i H3K4diMe, które nie są obserwowane w komórkach pochodzenia szpikowego.
Prawdopodobnie zmiany ekspresji genów VSMC mogą być spowodowane przez cholesterol zawarty w blaszkach miażdżycowych ponieważ w warunkach hodowli VSMC zaobserwowano wzrost ekspresji genów prozapalnych, obniżenie ekspresji markerów VSMC oraz ekspresję markerów makrofagów i pojawienie się zdolności do fagocytozy, która była jednak zmniejszona w stosunku do klasycznych makrofagów. Powyższe obserwacje wykazywały zależność od KLF4.

Wydaje się także, że prawidłowa budowa macierzy zewnątrzkomórkowej (ECM) wpływa hamująco na zmianę ekspresji genów w komórkach mięśni gładkich poprzez utrzymywanie w stanie skurczu, co wpływa na niższą wrażliwość na mitogeny. Jednak zaburzenie struktury ECM m.in. pod wpływem działania MMP z makrofagów i zmienionych VSMC sprzyja migracji i wspomnianym zmianom.
Obecnie strategie leczenia i zapobiegania rozwojowi miażdżycy skupiają się na lekach obniżających stężenie cholesterolu we krwi m.in. statyn. Postępy w badaniach nad patogenezą komórkową miażdżycy jakie miały miejsce w ostatnich latach, pozwalają stopniowo myśleć o poszukiwaniu leków wpływających na funkcję, utrzymanie pozytywnego fenotypu komórek oraz zmniejszyć reakcję zapalną, aby w ten sposób zabezpieczyć przed groźnymi dla życia powikłaniami takimi jak zawał serca czy udar mózgu, a także poprawić jakość życia pacjentów.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
KOMENTARZE