

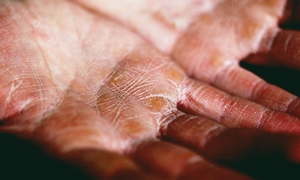
Kończynowy zespół złuszczającej się skóry (acral peeling skin syndrome) – to bardzo rzadka choroba skóry o dziedziczeniu autosomalnym recesywnym, spowodowana powstaniem mutacji genu TGM5 zlokalizowanego na chromosomie 15q15. Prawdopodobieństwo wystąpienia tej choroby u kolejnego dziecka wynosi 25%. Pierwsze objawy choroby pojawiają się we wczesnym dzieciństwie. Schorzenie to charakteryzuje się powstawaniem wiotkich, nietrwałych pęcherzy oraz powierzchownym złuszczaniem się naskórka rąk i stóp. Pojawiające się złuszczanie naskórka jest bezbolesne. Powstałe zmiany goją się bez pozostawania blizn, lecz czasami obserwuje się resztkowy rumień. Zaostrzenie objawów następuje w okresie podwyższonej temperatury otoczenia i wilgotności, a także po drobnych urazach. Przeprowadzone analizy ultrastrukturalne warstwy zewnętrznej naskórka wskazują, że połączenia pomiędzy komórkami ziarnistymi a warstwą rogową są nieścisłe. Schorzenie to powoduje obniżenie jakości życia z powodu braku skutecznego leczenia. Wartym podkreślenia faktem jest to, że wiele przypadków kończynowego zespołu złuszczającej się skóry jest błędnie rozpoznawanych jako pęcherzowe oddzielanie naskórka. Z dostępnych danych wynika, że nawet 35-50% diagnoz jest błędnych! By zmniejszyć łuszczenie się skóry, używa się emolientów. Obecnie nie ma informacji u skutecznym leczeniu kończynowego zespołu złuszczającej się skóry. Stosuje się leczenie objawowe oraz zapobiegawcze. W celu zapobiegania pojawianiu się nowych zmian chroni się skórę przed przegrzaniem, większą wilgotnością oraz urazami powstałymi na skutek nacisku i tarcia. Stosowanie antyperspirantów oraz zasypek ogranicza nadmierną potliwość, jednocześnie zmniejszając prawdopodobieństwo pojawienia się nowych zmian skórnych.
Wrodzone pęcherzowe oddzielanie się naskórka (epidermolysis bullosa, EB) – to choroba genetyczna, która charakteryzuje się nadmierną podatnością skóry oraz błony śluzowej na oddzielanie się od znajdujących się pod nimi tkanek w wyniku urazów mechanicznych. U osób chorych często pojawiają się również inne zmiany skórne, tj. prosaki, nadżerki, zanik płytek paznokciowych czy bliznowacenie. Ponadto objawy chorobowe występują ze strony innych narządów i układów (m.in. osteopenia, wodonercze, uszkodzenie przewodów słuchowych czy rogówki). Dziedziczenie zależy od rodzaju mutacji, genu odpowiedzialnego za EB i lokalizacji w genie (autosomalny recesywny oraz autosomalny dominujący). Dziedziczenie jest niezależne od płci. W przypadku niektórych podtypów tej choroby wzrasta znacząco ryzyko rozwoju nowotworu. U jednych zmiany mają charakter łagodny, zaś u innych choroba przebiega bardzo ciężko, prowadząc nawet do śmierci w pierwszych miesiącach życia. Wyróżniamy cztery odmiany wrodzonego pęcherzowego oddzielania się naskórka ze względu na umiejscowienie zmian chorobowych w tkance skórnej:
* epidermolysis bullosa simplex (EBS) – zmiany zlokalizowane w obrębie naskórka. Jest to postać najłagodniejsza i najczęściej występująca. Jej charakterystycznymi objawami są pęcherze powstałe w miejscu pocierania, zwłaszcza na rękach i stopach,
* dystrophic epidermolysis bullosa – zmiany zlokalizowane w skórze właściwej. Postać dystroficzna jest spowodowana przez mutacje, które wpływają na produkcję kolagenu. W ciężkiej postaci EB powstają różne komplikacje, które wpływają na możliwość wyleczenia. Najczęstsze powikłania to: niedokrwistość, świąd, ból oraz zakażenia,
* epidermolysis bullosa junctionalis – zmiany pojawiają się w błonie podstawnej, która jest umiejscowiona pomiędzy naskórkiem a skórą właściwą. Schorzenie to pojawia się już przy urodzeniu i ma zazwyczaj cięższy przebieg niż postać zwykła zlokalizowana w obrębie naskórka. Ten typ ma wpływ na laminę oraz kolagen.
* zespół Kindlera – zmiany obserwuje się w różnych częściach granicy skórno-naskórkowej. Jest to najrzadsza postać, trudna do zdiagnozowania. Powstaje w wyniku mutacji w genie FERMT1, które powodują pojawianie się pęcherzy, zanikanie naskórka oraz opóźnione gojenie się skóry.
Każda wspomniana odmiana tego schorzenia dzieli się na podtypy. Wyróżnia się również podział ze względu na postać nabytą oraz wrodzoną.
Acrodermatitis enteropathica to zaburzenie powstające na skutek niedoboru cynku. Częstotliwość występowania szacowana jest na ok. 1:500000. U chorych brakuje zdolności wchłaniania cynku z przewodu pokarmowego. Choroba jest diagnozowana we wczesnym dzieciństwie oraz kilka dni po urodzeniu. Częstość występowania choroby jest nieznana. Do charakterystycznych objawów zalicza się zmiany skórne zlokalizowane w okolicy otworów ciała – wokół ust, odbytu oraz w okolicy kończyn (dłonie, podeszwy stóp, palce rąk i stóp). Objawy typowe dla acrodermatitis enteropathica to: zmiany wypryskowo-wysiękowe, łuszczenie oraz strupy. Często dochodzi do powstawania licznych, trudnych do wyleczenia nadżerek, które się podatne na wtórne zakażenia gronkowcowe i grzybicze. Pojawiające się zapalenie skóry jest bardzo podobne do zmian występujących w łuszczycy. Co więcej, oprócz objawów skórnych obserwuje się wypadanie włosów oraz zakażenie wału paznokciowego. Należy podkreślić, że prawie u każdej osoby z niedoborem cynku obserwuje się wypadanie włosów i zanokcicę. Ciężkie objawy związane z przewlekłym niedoborem cynku to: zaburzenie wzrostu, upośledzenie smaku, nadwrażliwość na światło oraz zaburzenia ze strony układu nerwowego. Późne rozpoznanie choroby i związany z tym brak leczenia powodują, że większość pacjentów umiera w młodym wieku albo choroba przebiega bardzo ciężko. Leczenie polega na doustnej suplementacji cynku. Terapia niezwykle szybko przynosi pozytywne efekty – zmiany na skórze zaczynają się goić i ustępują w ciągu tygodnia. W czasie dojrzewania oraz w czasie ciąży należy przyjmować większe dawki leku.




KOMENTARZE