

Ewolucja pomieszczeń czystych, technologii izolacji oraz systemów barierowych
Ewolucja pomieszczeń czystych oraz systemów barierowych odegrała kluczową rolę w wytwarzaniu produktów sterylnych. Za początek pomieszczeń czystych można uznać opracowanie wysokowydajnego systemu filtracji cząstek stałych (HEPA) na początku lat 40. XX w. podczas badań nad bronią atomową w ramach Projektu Manhattan. [1]
Technologia izolacji podobnie zrodziła się z badań nad bronią atomową prowadzonych w czasie II wojny światowej. Niemal od początku produkcji wyrobów sterylnych wiadomo było, że głównym źródłem zanieczyszczeń jest operator. Ta świadomość doprowadziła do bardzo wczesnego zastosowania izolatorów w produkcji sterylnej, jednak wprowadzenie na rynek i upowszechnienie filtrów HEPA w latach 50. XX w. umożliwiło utworzenie całych pomieszczeń czystych skoncentrowanych głównie na kontroli cząstek stałych. Postęp ten tymczasowo przyćmił zastosowanie technologii izolatorów w przemyśle farmaceutycznym, ale wraz z rozwojem branży i nasilającymi się wymaganiami konsumentów dotyczącymi poprawy jakości produktów, potrzeba ściślejszej kontroli zanieczyszczeń i sterylności stawała się coraz bardziej oczywista. W 1967 r. firma LaCalhene wprowadziła na rynek swój pierwszy izolator i stała się pionierem w technologii izolacji, zwłaszcza w Europie. Na początku lat 70. XX w. wprowadzenie Dobrych Praktyk Produkcyjnych (GMP) zrewolucjonizowało obraz produkcji farmaceutycznej, przynosząc znaczną poprawę procesów kontroli zanieczyszczeń. [2]
Przyszłość aseptycznego przetwarzania i przygotowania
Obecnie techniki aseptyczne wykorzystują zwykle urządzenia z przepływem laminarnym lub technologie barierowe w celu zmniejszenia ryzyka zanieczyszczenia mikrobiologicznego i cząstkami stałymi.
Urządzenia z przepływem laminarnym
Komory laminarne
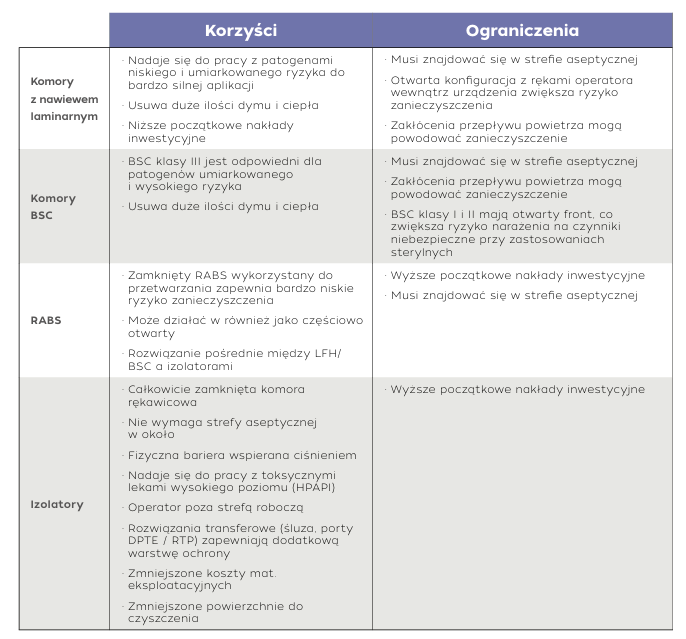
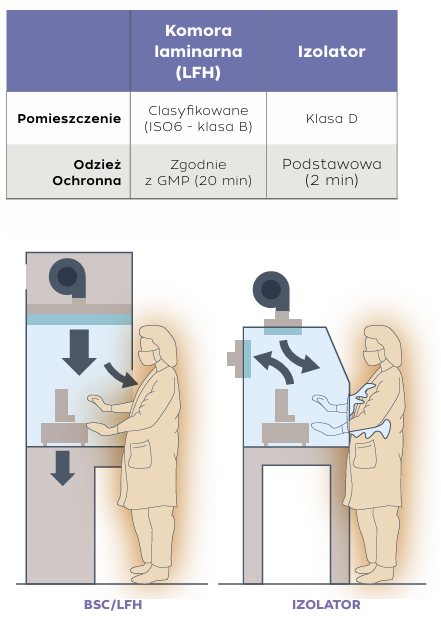
Komory laminarne (LFH) wykorzystują filtrację HEPA i jednokierunkowy, laminarny przepływ powietrza w celu ochrony produktów na powierzchni roboczej przed zanieczyszczeniem cząstkami stałymi. To niezmącone powietrze jest kierowane na powierzchnię roboczą, aby fizycznie owiewać wnętrze komory i zapobiegać przedostawaniu się cząstek stałych z otaczającego środowiska. Ponieważ nawiewy z przepływem laminarnym są systemami otwartymi, muszą być używane w pomieszczeniu czystym, zwykle klasy B. Komory laminarne z nawiewem poziomym kierują przepływ powietrza równolegle do powierzchni roboczej, podczas gdy w pionowych komorach powietrze porusza się pionowo w dół. Zarówno w konfiguracjach poziomych, jak i pionowych powietrze jest zwykle wydmuchiwane z przodu komory, co sprawia, że komory laminarne nie nadają się do zastosowań wymagających ochrony operatora
Komory bezpieczeństwa biologicznego
Istnieją trzy rodzaje komór bezpieczeństwa biologicznego (BSC): klasa I, klasa II i klasa III. Chociaż wszystkie trzy typy komór bezpieczeństwa biologicznego zapewniają ochronę personelu i środowiska, tylko komory klasy II i klasy III zapewniają ochronę produktu. Wykorzystują one filtry HEPA do usuwania mikroskopijnych zanieczyszczeń z powietrza, które podlega recyrkulacji i trafia z powrotem do laboratorium. BSC klasy II nadają się do zastosowań związanych z produktami i mikroorganizmami niskiego, umiarkowanego i wysokiego ryzyka. BSC klasy I i II są systemami otwartymi, które nie stanowią całkowitej bariery między produktem a operatorem. BSC klasy III to systemy zamknięte, które chronią operatorów, produkt i otaczające środowisko. Ze względu na wysoce skomplikowane działanie i wysokie koszty BSC klasy III, są one idealne do pracy z wysoce zakaźnymi czynnikami mikrobiologicznymi, a ich przydatność w zastosowaniach farmaceutycznych jest ograniczona.
Technologie barierowe
Systemy barier o ograniczonym dostępie (RABS)
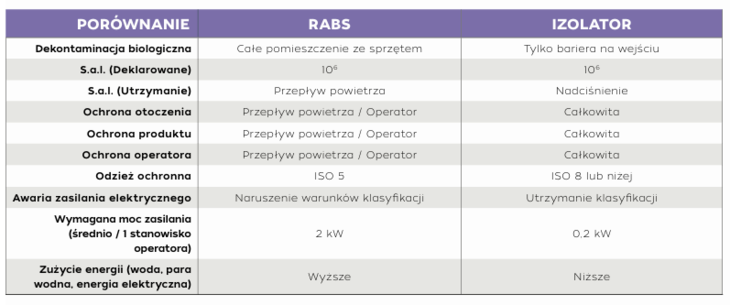
Systemy RABS utrzymują nadciśnienie wewnątrz obudowy, zapobiegając przedostawaniu się zanieczyszczeń i zapewniając fizyczną barierę między operatorem a produktem. Dostępne są w konfiguracjach otwartych i zamkniętych oraz typu aktywnego lub pasywnego. Otwarte RABS zapewniają tylko częściową barierę. W wersji pasywnej nie zawierają one wewnętrznego systemu filtracji HEPA, natomiast wykorzystują systemy laminarnego przepływu powietrza w samym pomieszczeniu produkcyjnym. W wersji aktywnej zawierają zaś niezależny system przepływu laminarnego. Systemy RABS typu otwartego zazwyczaj muszą być zlokalizowane w pomieszczeniu czystym. Zarówno w aktywnych, jak i pasywnych otwartych RABS obszar wewnątrz musi być klasy A, a otaczający go obszar jest sklasyfikowany jako klasa B. Zamknięte RABS działają podobnie do technologii izolatorów, ponieważ zapewniają kompletną barierę, a zatem mają mniejsze ryzyko zanieczyszczenia. Główna różnica między zamkniętymi RABS a izolatorami polega na tym, że zamknięte RABS nie mają zautomatyzowanego cyklu biodekontaminacji z wykorzystaniem H2O2 lub innych metod.
Izolatory
Izolatory zapewniają najwyższy poziom ochrony i elastyczność w aseptycznym przetwarzaniu. Tworząc hermetyczną barierę między produktem, procesem i personelem, izolatory skutecznie eliminują ryzyko zanieczyszczenia mikrobiologicznego i cząstkowego. Ta skrupulatna separacja chroni integralność produktu, ustanawiając nowy standard zapewnienia jakości w produkcji farmaceutycznej. Są one idealnym rozwiązaniem do pracy z silnymi lub toksycznymi substancjami, ponieważ minimalizują ryzyko narażenia operatora na ich działanie i zanieczyszczenia produktu. Dzięki całkowitemu zamknięciu procesu aseptycznego w szczelnym środowisku, proces jest chroniony przed zanieczyszczeniami ze strony operatora. Poprawia to kontrolę jakości produktu, skraca czas przestojów i ułatwia śledzenie źródeł zanieczyszczeń wykrytych w testach sterylności. Ponieważ technologia izolacji chroni również operatora, może być wykorzystywana w szerszym zakresie zastosowań, w tym w aplikacjach związanym z pracą z toksycznymi chemikaliami lub lekami, silnymi i bardzo silnymi aktywnymi składnikami farmaceutycznymi (HPAPIs), a także w sterylnym napełnianiu produktów i przetwarzaniu aseptycznym.

Skuteczniejsza kontrola zanieczyszczeń dzięki systemom barier
Ponieważ zanieczyszczenia są kluczowym problemem w technikach aseptycznych, należy przede wszystkim rozważyć technologię, która może zmniejszyć ryzyko zanieczyszczenia mikrobiologicznego i cząsteczkowego. Komory laminarne i BSC zapewniają tylko częściowe oddzielenie od operatora i procesu. Nawet gdy są one używane w tradycyjnych pomieszczeniach czystych, to stanowią niewielką realną barierę przed wnikaniem zanieczyszczeń. Aby poprawić kontrolę zanieczyszczeń w aseptycznej produkcji i przetwarzaniu, konieczne jest całkowite oddzielenie operatora od procesu. Jest to szczególnie ważne w przypadku bardzo wrażliwych aplikacji. Zaawansowane przetwarzanie aseptyczne (AAP) to krytyczny proces, który wymaga najwyższego poziomu kontroli zanieczyszczeń. W związku z tym techniki AAP wymagają użycia prawdziwego systemu barierowego. [3]
Zgodnie z „European Pharmaceutical Review” pełna kontrola wnikania osiągnięta poprzez zastosowanie izolatora lub niektórych systemów RABS zapewnia kontrolę zanieczyszczeń powyżej poziomu możliwego do uzyskania w tradycyjnym pomieszczeniu czystym. [4] Systemy barierowe zapewniają fizyczne bariery, które oddzielają produkt od otaczającego środowiska, zmniejszając możliwość przedostania się zanieczyszczeń unoszących się w powietrzu do strefy aseptycznej i obejmują systemy izolatorów oraz RABS.
Rosnąca popularność technologii barierowych
Przepisy dotyczące Dobrych Praktyk Produkcyjnych ewoluowały od czasu ich wprowadzenia. Aktualne wytyczne Dobrych Praktyk Produkcyjnych (cGMP) obejmują zalecenia dotyczące zapewnienia wyższego poziomu kontroli zanieczyszczeń podczas aseptycznego przygotowywania i przetwarzania produktów. Rygorystyczne wytyczne określone przez organy regulacyjne, takie jak Amerykańska Agencja ds. Żywności i Leków (FDA) i Unia Europejska (UE), stanowią podstawę zapewnienia bezpieczeństwa i sterylności produktów. Wytyczne te określają krytyczne parametry i procedury, których należy przestrzegać w całym procesie produkcyjnym i które odgrywają kluczową rolę w aseptycznym przygotowaniu i przetwarzaniu.
Wymagany poziom kontroli zanieczyszczeń różni się w zależności od konkretnych operacji aseptycznego przygotowania i przetwarzania. Zdefiniowano różne stopnie operacji, z których każdy ma własny zestaw wymagań. Na przykład środowiska klasy A charakteryzują się najwyższym poziomem czystości i są zarezerwowane dla krytycznych operacji sterylnych. Klasa B jest zarezerwowana dla otoczenia środowisk, które są często wykorzystywane do aseptycznego przygotowania, a w wybranych przypadkach – również napełniania niektórych produktów klasy A. Klasy C i D są idealne do realizacji niekrytycznych etapów produkcji.
To, czy w danej aplikacji należy wykorzystywać pomieszczenie czyste z komorą laminarną lub BSC, czy raczej technologię barierową, taką jak RABS lub izolator, zależy od poziomu pożądanej kontroli zanieczyszczeń i wymogów prawnych. Choć może wydawać się dziwne, że przyszłością technik aseptycznych jest technologia istniejąca od samego początku, zalety izolatorów w produkcji farmaceutycznej są oczywiste. W sierpniu 2022 r. opublikowano Załącznik 1 do obowiązujących w UE Dobrych Praktyk Produkcyjnych dla produktów leczniczych stosowanych u ludzi i weterynaryjnych produktów leczniczych, w którym przyjęto nowe podejście do zapewnienia sterylności. W załączniku tym zaproponowano całościowe podejście do zapobiegania zanieczyszczeniom w produkcji leków sterylnych, a także leków, które nie wymagają produkcji sterylnej. W poprzednich wersjach obowiązujących w UE cGMP dla produkcji medycznej w przypadku produkcji farmaceutycznej z zastosowaniem technik aseptycznych odpowiednim rozwiązaniem były środowiska klasy A (pomieszczenia czyste). Tradycyjnie środowiska klasy A są otoczone kolejnymi kontrolowanymi środowiskami od klasy B do C, przy czym każde oddzielne środowisko ma własne standardowe procedury operacyjne (SOP) zapewniające to, że zanieczyszczenie mikrobiologiczne i cząsteczkowe w powietrzu mieści się w koniecznych granicach.
Opublikowanie Załącznika 1 spowodowało, że pomieszczenia czyste nie są już preferowaną metodą kontroli zanieczyszczeń, a ich użycie musi być uzasadnione. Nowe zalecenia UE wymagają stosowania technologii barierowych, w tym RABS lub izolatorów. Chociaż wymagania Załącznika 1 mają obecnie zastosowanie tylko do rynku europejskiego, to w najbliższej przyszłości również w innych światowych regulacjach prawdopodobnie zostaną przyjęte podobne zmiany służące uzyskaniu wyższego poziomu zapewnienia sterylności (SAL).

Zalety technologii izolatorów w sterylnej i niesterylnej produkcji farmaceutycznej
Wiele produktów sterylnych, w tym produktów biologicznych, nie może być sterylizowanych na ostatnim etapie produkcji. W związku z tym niezbędne jest zapewnienie sterylności produktu podczas produkcji i przetwarzania. Zalety izolatorów są następujące:
• zdolność izolatorów do utrzymania wyższego stopnia sterylności poprzez wyeliminowanie ryzyka kontaktu z ludźmi podczas przetwarzania i przygotowywania,
• zmniejszone ryzyko zanieczyszczenia i wyższa jakość produktu dzięki zamkniętemu i kontrolowanemu środowisku izolatora,
• zwiększone bezpieczeństwo operatora poprzez zminimalizowanie narażenia na niebezpieczne materiały i zmniejszenie ryzyka zanieczyszczenia krzyżowego,
• zwiększona produktywność i wydajność dzięki możliwości automatyzacji procesów w izolatorze,
• większa zgodność z wymogami prawnymi i standardami branżowymi w zakresie aseptycznego przetwarzania i przygotowywania produktów,
• znaczna redukcja kosztów związanych z eksploatacją pomieszczeń czystych (czyszczenie, fartuchy, powierzchnia, koszty bieżące),
• w porównaniu do pomieszczeń czystych izolatory zapewniają mniejsze środowisko wymagające kontroli.
Międzynarodowe Stowarzyszenie Inżynierów Farmaceutycznych (ISPE) opublikowało niedawno ankietę, w której odnotowano rosnące wykorzystanie systemów barierowych, takich jak izolatory i RABS, do aseptycznego napełniania. [5] Chociaż trend ten doprowadził do „niemal całkowitego wyginięcia” tradycyjnych pomieszczeń czystych do aseptycznego napełniania, nadal istnieją podmioty, które nie wdrożyły żadnej formy technologii barierowej. Podczas konferencji ISPE Aseptic Barrier 2020 paneliści FDA stwierdzili, że zakłady, które nadal korzystają z tradycyjnych pomieszczeń czystych klasy A, odnotowują większe wskaźniki wycofywania produktów z rynku, a zatem podlegają wyższemu poziomowi kontroli. Izolatory przyczyniają się do optymalizacji procesów produkcyjnych. Zmniejszając potrzebę ręcznych interwencji i minimalizując prawdopodobieństwo zanieczyszczenia, przyspieszają one cykle produkcyjne i zwiększają wydajność.
Izolatory nadal ewoluują wraz z postępem technologicznym, stając się coraz bardziej wyrafinowane i przyjazne dla użytkownika. Najnowocześniejsze systemy automatyzacji i kontroli umożliwiają precyzyjne monitorowanie i dostosowywanie parametrów procesu, zapewniając stałą jakość produktu i minimalizując ryzyko błędu ludzkiego. Ponadto mechanizmy transferu izolatorów (systemy RTP), które zostały opracowane wkrótce po opracowaniu izolatorów w celu rozwiązania problemu bezpiecznego transferu, zapewniają skuteczne wkładanie i wyjmowanie materiałów, w tym wyrobów szklanych, materiałów eksploatacyjnych, mediów, produktów itd., bez naruszania sterylnej szczelności. Przykładem rozwiązania RTP jest system Getinge DPTE®, który był pierwszym w branży systemem szybkiego transferu i podlega certyfikacji pod kątem wyeliminowania zagrożeń mikrobiologicznych i cząstek stałych podczas transferu materiałów, odpadów lub produktów. Umożliwia on transfer materiału do izolatora, ale także ewakuację materiałów na zewnątrz, w tym poprzez podwójny worek na odpady stałe i ciekłe, jak w przypadku testów sterylności. Kompletny system DPTE® ma potwierdzoną szczelność i jest zatwierdzony do aseptycznego transferu w całym łańcuchu produkcji farmaceutycznej.
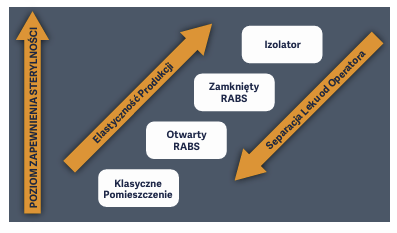
Przewiduje się, że w miarę rozwoju aseptycznego przetwarzania i przygotowywania produktów, izolatory będą odgrywać jeszcze ważniejszą rolę w kształtowaniu sektora. Ich zdolność do zapewnienia kontroli zanieczyszczeń, zwiększenia bezpieczeństwa operatora i optymalizacji wydajności rewolucjonizuje produkcję farmaceutyczną, ostatecznie prowadząc do wytwarzania produktów najwyższej jakości i gwarancji bezpieczeństwa pacjentów.
Testy sterylności
Zanieczyszczenie farmaceutyków i innych produktów jest zazwyczaj niedostrzegalne gołym okiem, co często uniemożliwia konsumentowi określenie bezpieczeństwa i skuteczności produktu leczniczego. Chociaż wszystkie testy kontroli jakości, takie jak test sterylności, podlegają określonym ograniczeniom, to test ten pozostaje istotnym elementem kompleksowego planu kontroli mającego na celu zapobieganie skażeniu mikrobiologicznemu produktów leczniczych, które muszą być sterylne. Testowanie sterylności jest ostatnią czynnością przeprowadzaną na sterylnych produktach gotowych, w tym antybiotykach w formie proszków, wstrzykiwanych płynach i produktach okulistycznych, i jest ono wymagane przed dopuszczeniem partii do obrotu. Testy te są przeprowadzane w izolatorze lub komorze laminarnej/BSC w celu oceny produktów pod kątem obecności mikroorganizmów lub innego przetestowania jakości partii produktów sterylnych. Ponieważ w wyniku tych testów produkt nie nadaje się do użytku, są one przeprowadzane na małych próbkach całej partii lub jej opakowania przy użyciu różnych metod, takich jak filtracja membranowa, bezpośrednia inokulacja lub metody oparte na wzroście, np. test sterylności przeprowadzony w ramach końcowej kontroli jakości partii 2000 fiolek wiąże się z wyborem 20 fiolek do przeprowadzenia testu.
BSC/LFV vs. izolator
Najskuteczniejszym sposobem zapewnienia sterylności aseptycznych procesów napełniania i procesów końcowych jest zastosowanie technologii izolatorów. Izolatory zapewniają wysoką skuteczność zapobiegania zanieczyszczeniu mikrobiologicznemu i cząsteczkami podczas procesów aseptycznych, w tym produkcji sterylnych leków i produktów biologicznych, takich jak szczepionki, terapie genowe i leki na bazie białek. Izolator do testów sterylności wyposażony w port szybkiego transferu (RTP) w połączeniu z podwójnym workiem na odpady pozwala na bezpieczne i wydajne usuwanie odpadów płynnych i stałych. RTP zapewnia bezpieczną ewakuację materiału z wnętrza komory izolatora, przy jednoczesnym zachowaniu integralności izolatora, eliminując w ten sposób ryzyko zanieczyszczenia próbki przez środowisko, a tym samym – zmniejszając liczbę fałszywych wyników dodatnich. Chociaż pozostałości nadtlenku wodoru po automatycznej dekontaminacji biologicznej w izolatorze mogą zwiększać ryzyko fałszywych wyników ujemnych, problem ten można ograniczyć za pomocą odpowiednich SOP-ów, które zapewniają zweryfikowany proces wykluczający pozostałości H2O2. [6] Zgodnie z USP Testy Sterylności „systemy izolatorów chronią testowane artykuły i materiały do testów sterylności przed zanieczyszczeniem podczas pracy z zachowaniem aseptyczności”. [7]
Izolatory mogą być zaprojektowane w sposób zapewniający ochronę produktów biologicznych i innych produktów wrażliwych na zanieczyszczenie (poprzez nadciśnienie) albo ochronę operatora przed toksycznymi, cytotoksycznymi i wysoce silnymi aktywnymi składnikami farmaceutycznymi podczas pracy (poprzez podciśnienie). Zmniejszają one ryzyko nieprawidłowych manipulacji przez zapewnienie tego, że manipulacje produktem są przeprowadzane w kontrolowanym, sklasyfikowanym obszarze.

Podsumowanie
Przestrzegając wytycznych określonych w przepisach, wdrażając skuteczne Strategie Kontroli Zanieczyszczeń (CCS) i wybierając odpowiedni sprzęt, producenci z branży farmaceutycznej mogą zapewnić najwyższy poziom kontroli zanieczyszczenia mikrobiologicznego i cząsteczkowego. To z kolei gwarantuje wysoką jakość produktów, bezpieczeństwo pacjentów i zgodność z przepisami. Ryzyko zanieczyszczenia stanowi poważne zagrożenie dla jakości produktu i bezpieczeństwa pacjentów, co wymaga wdrożenia skutecznych strategii kontroli zanieczyszczeń. Strategie te obejmują szereg działań, w tym szkolenie personelu, monitorowanie środowiska i stosowanie odpowiedniej technologii barierowej. Izolatory zapewniają wyższy poziom ochrony przed skażeniem niż urządzenia z przepływem laminarnym, takie jak komory laminarne, szafy bezpieczeństwa biologicznego oraz RABS. Zarówno amerykańskie, jak i unijne wytyczne cGMP dotyczące wytwarzania produktów leczniczych stosowanych u ludzi i w weterynarii szczegółowo opisują korzyści płynące ze stosowania technologii izolatorów w zapobieganiu zanieczyszczeniom. [8,9]
Zastosowania technologii izolatorów:
• napełnianie i wykańczanie małych partii,
• testy sterylności lub kontrola jakości (filtracja membranowa, bezpośrednia inokulacja),
• transfer lub biodekontaminacja,
• mieszanie składników (żywienie pozajelitowe TPN, wlewy dożylne, rekonstytucja cytotoksyków),
• hodowla komórkowa, terapia komórkowa i genowa,
• medycyna spersonalizowana,
• przetwarzanie aseptyczne (montaż procesów, przygotowanie części itp.),
• transfer i przepakowywanie sterylnych komponentów, korków, kapsli, itp.,
• izolator do napełniania reaktora lub zbiornika,
• montaż i przygotowanie wyrobów medycznych.

Artykuł przygotowano w oparciu o white paper „Comparing Laminar Flow Hood and Barrier Technology in Aseptic Processing”.
Autor: Grzegorz Cacko, Life Science Sales Manager Getinge Polska Sp. z o.o.




KOMENTARZE