


Właśnie dlatego, mając do dyspozycji nanoporową technologię sekwencjonowania naturalnym dla nas było skupić się także na odczytaniu i zbadaniu sekwencji genomów koronawirusa SARS-CoV-2. Przypadki tych wirusów są identyfikowane przez nasze laboratorium diagnostyki molekularnej, a następnie sekwencjonowane w laboratorium badawczo-rozwojowym i analizowane w dziale bioinformatyki. Pozwoliło nam to zasilić sekwencjami koronawirusów z Polski otrzymanymi w genXone międzynarodową bazę GISAID, a oprócz tego przeprowadziliśmy analizę bioinformatyczną wszystkich 80 000 opublikowanych do tej pory sekwencji koronawirusa pod kątem jego zmian w genomach.
Ewolucja wirusa dokonuje się w nas. Koronawirus wnika do komórek w naszym organizmie, a następnie namnaża się poprzez przepisywanie swej sekwencji RNA bezpośrednio na nowe kopie genomu lub jego fragmentów. W pierwszym momencie po wniknięciu do ludzkiej komórki wykorzystuje on działające tam mechanizmy, jednak do właściwej replikacji używa już własnych, zakodowanych w swoim genomie białek kompleksu RTC oraz enzymu RdRP (w genomie koronawirusa sekwencja ta nosi nazwę nsp12 i jest jednym z 16 białek zakodowanych w ORF rep1b na początku sekwencji jego genomu). Jakość działania, czyli dokładność RdRP i towarzyszących mu enzymów w głównej mierze decyduje o tym, jak często powstają nowe pojedyncze mutacje w kolejnych kopiach wirusa. Rzadziej zdarzają się mutacje spontaniczne powstające przez naturalny kwantowo-termodynamiczny rezonans struktur chemicznych lub mutacje indukowane (np. promieniowanie UV).
Choć obecnie atakujący nas koronawirus SARS-CoV-2 mutuje zdecydowanie wolniej niż wirus grypy bądź wirus HIV, to mimo wszystko zmiany w jego genomie postępują. U poszczególnych koronawirusów obserwujemy różnice względem pierwotnego wirusa z Wuhan w dziesięciu i więcej nukleotydach spośród pełnej sekwencji jego RNA. Uwzględniając wyniki sekwencjonowania dla wirusa SARS-CoV-2 z całego świata, średnia (mediana) różnic w sekwencji nukleotydów wynosi 8. W samej Polsce ta średnia wynosi 10 nukleotydów różnicy od sekwencji genomu pierwotnego wirusa z Wuhan, czyli więcej niż na świecie. Jest tak, gdyż wirus dotarł do nas trochę później i zdążył w tym czasie zmutować.
Polskimi rekordzistami okazały się dwa genomy wirusa: z województwa łódzkiego oraz pomorskiego. Próbka z województwa łódzkiego została zsekwencjonowana w laboratorium genXone i wykazała 18 różnic w stosunku do pierwotnego wirusa z Chin. Podobnie próbka z województwa pomorskiego posiada 18, lecz odmiennych, wariantów. Ze względu na odmienne metody raportowania danych przez kraje ciężko ustalić niezaprzeczalnego rekordzistę na skalę światową. Jednakże obserwujemy przypadki posiadające 20 lub więcej zmienionych nukleotydów.
Obserwowana w analizie prędkość pojawiania się mutacji u obecnego koronawirusa oznacza jedną, nową zmianę nukleotydu z prawie każdym kolejnym zarażeniem. Ten fakt daje nam pewną przewagę – jest to możliwość prześledzenia łańcucha zakażeń wśród ludzi jedynie przy pomocy analiz sekwencji wirusowych genomów.
Historia z mutacjami koronawirusa na tym się nie kończy. Otóż wspomniany enzym RdRP odpowiedzialny za kopiowanie koronawirusów znany jest z tego, iż potrafi przeskoczyć z kopiowanej nici RNA na nić sąsiadującą, co w efekcie, raz na jakiś czas, powoduje powstanie rekombinowanego genomu, czyli sekwencji powstałej przez połączenie dwóch fragmentów źródłowych, np dwóch genomów koronawirusów SARS-CoV-2 lub, w przypadku koinfekcji, dwóch genomów koronawirusów z różnych szczepów. Takowych szczepów zarażających człowieka poznano od lat 60. przynajmniej siedem. Właśnie takie zdarzenia rekombinacji przyczyniają się do ewolucji koronawirusów, które charakteryzują się dość dużą zmiennością, intensywnie rozwijając się w organizmach nietoperzy, łuskowców i innych ssaków.
Ewolucję obecnego koronawirusa SARS-CoV-2 możemy poznać i zrozumieć dzięki międzynarodowym inicjatywom sekwencjonowania. Inicjatywa GISAID promuje szybkie współdzielenie danych dotyczących wszystkich wirusów grypy oraz ostatnio koronawirusa powodującego COVID-19. Współdzielone są dane sekwencji genetycznych wirusa oraz związane z nimi dane kliniczne i epidemiologiczne, geograficzne, a w przypadku wirusów odzwierzęcych także gatunkowe. Celem inicjatywy jest zrozumienie, w jaki sposób wirusy ewoluują i rozprzestrzeniają się podczas pandemii.
Najwięcej koronawirusów na świecie zsekwencjonowano w tym roku w Wielkiej Brytanii (pow. 30 000), na drugim miejscu jest USA (pow. 17 000), dalej Australia i Hiszpania, każda z ponad 2700 sekwencjami wirusa. W Polsce inicjatywę GISAID w gromadzeniu bazy genomów koronawirusa oprócz genXone wsparło również sześć innych instytucji. W wyniku tego z końcem lipca dostępnych jest w bazie 115 sekwencji koronawirusów z Polski. Docelowo genXone planuje udostępnić do bazy wyniki sekwencjonowania wszystkich pozytywnych próbek koronawirusowych zidentyfikowanych w swoim laboratorium.
Pojedynczy genom wirusa niesie ze sobą informację o historii jego mutacji. Opisana wyżej częstość mutowania oraz dokładne dane z sekwencjonowania pozwalają odtworzyć ścieżkę kolejnych zakażeń. Porównane genomy wielu wirusów pozwalają odtworzyć drzewo genealogiczne, na którym rozwidlenia powstają w momencie kolejnych mutacji wirusa. Śledzenie krawędzi pozwala poznać kierunki przepływu wirusów pomiędzy regionami świata.
Co interesujące, proces ewolucji koronawirusa można zaobserwować bezpośrednio w komórkach zbadanych osób. Zsekwencjonowane w genXone próbki świadczą o występowaniu u jednego pacjenta przynajmniej dwóch różnych wariantów wirusa: z nową mutacją i bez niej. Ich obserwowane proporcje sięgają 35%, zidentyfikowaliśmy przynajmniej dwie takie próbki.
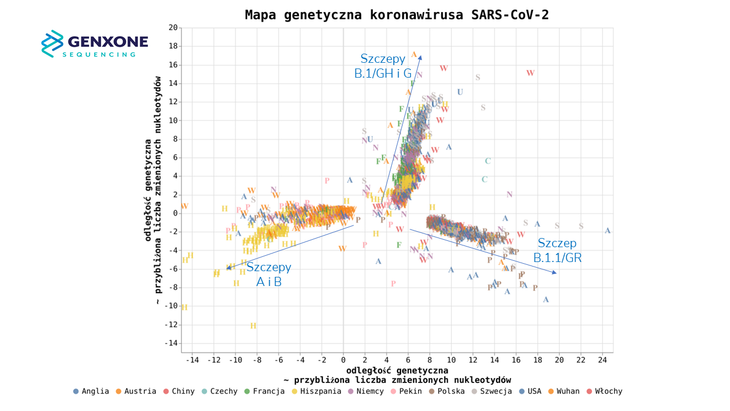
Interaktywna mapa genetyczna koronawirusa na próbce danych z wybranych krajów. Odstępy na mapie przybliżają odległość genetyczną między próbkami (PCoE metodą Sammona). Dane GISAID EpiCOV z początku sierpnia 2020 r. Autor: Maciej Sykulski, genXone.
Dziś na świecie możemy wyróżnić gałęzie wirusa bliższe sekwencji z Wuhan, czyli szczep B i występujący w pozostałych regionach Chin szczep A. Warto odnotować, iż znaczący procent wirusów w Hiszpanii wywodzi się z chińskich gałęzi, co odróżnia Hiszpanię od ogólnych wyników europejskich, gdzie dominują dwie odmienne odnogi wirusa: szczepy B.1 oraz B.1.1. Właśnie te dwa ostatnie odniosły największy sukces w Europie i dopiero później trafiły na kolejne kontynenty. Szczepy B.1 oraz B.1.1 posiadają mutacje w sekwencji ORF1ab (4715L) i białku S (D614G), które zwiększają zakaźność wirusa. Ostatnie badania pokazują, że z tymi wariantami związana jest niestety także większa śmiertelność.
Sekwencje koronawirusów z woj. łódzkiego otrzymane w laboratorium genXone generalnie pasują do europejskich wzorców. Na przybliżonej mapie genetycznej wirusa widzimy przedstawicieli dwóch głównych szczepów w Europie: B.1/GH i B.1.1/GR i brak widocznych wirusów związanych bezpośrednio z Chinami. Dokładniejsze spojrzenie pozwala podejrzewać, iż w przypadku szczepu B.1/GH przybył on do woj. łódzkiego z Niemiec, Anglia jest mniej prawdopodobnym źródłem. W przypadku szczepu B.1.1/GR obecne są gałęzie wirusa, które krążyły już w marcu po Anglii, Austrii, Czechach, Niemczech i Polsce, więc nie można bez bardziej precyzyjnej analizy podać kierunku przepływu wirusa. Nie można wykluczyć, iż wirus przemieszczał się zarówno z zagranicy do Polski, jak i z Polski za granicę do wymienionych krajów.
Zaobserwowaliśmy ciekawą rzecz – otóż w woj. łódzkim zaobserwowaliśmy pięć próbek, które posiadają charakterystyczny wariant widoczny jedynie w czterech innych wynikach na całym świecie. Mutacja ta odnaleziona była jedynie w trzech próbkach w Anglii oraz w jednej w USA. Sekwencje genomów wirusów pokazują, iż jeden z pacjentów w Anglii był w kwietniu zarażony koronawirusem posiadającym tenże wariant. Następnie wirus mutant zawędrował do woj. łódzkiego, gdzie ewoluował niezależnie od dwóch pozostałych angielskich przypadków. Natomiast próbka z USA, wcześniejsza z marca br., jest odrębnym, wczesnym przypadkiem tej samej mutacji. Wariant ten może okazać się powracający lub rekombinowany, a także istotny dla zaraźliwości wirusa, ponieważ zmienia on aminokwas w białku S "Spike", który pozwala wnikać wirusowi w ludzkie komórki przez łączenie się z receptorem ACE2 osadzonym w błonie komórkowej.
Wykrywanie i poznawanie podobnych wariantów może mieć więc duże znaczenie dla efektywności terapii, poza tym wygląda na to, iż w najbliższym czasie pojawi się kilka szczepionek na koronawirusa. Jednak, czy wszystkie będą tak samo skutecznie na wszystkie szczepy wirusa? Co więcej, mutacje wirusa mogą wpływać na działanie szczepionek, a jego ewolucja może zostać ukierunkowana pod wpływem szczepień, selekcja wywarta na niego przez uodpornione komórki może spowodować powstawanie szczególnych mutantów, które mogą umykać działaniu szczepionki.
Sekwencjonowanie obecnego koronawirusa, poza wykrywaniem mutantów, daje jeszcze jedną przewagę: poznania drzewa zakażeń i jego cech charakterystycznych. Analizy naukowe z Australii, analizy śledzenia kontaktów w Hong Kongu, Izraelu oraz statystyczne analizy danych WHO z początku epidemii wykonane w Wielkiej Brytanii wskazują na to, iż wirus rozprzestrzenia się niejednorodnie, charakterystyczne jest tu zjawisko "superroznosicieli", tzn. szacowane jest, iż 80% zakażeń powodowane jest przez jedynie 10% zakażonych osób. Taki sposób rozchodzenia się wirusa nazywamy wysokim czynnikiem dyspersji. Dane genetyczne wirusów zsekwencjonowanych na świecie wydają się potwierdzać tę hipotezę. Dobra próba losowa genomów wirusa z Polski i świata daje szansę oszacować czynnik dyspersji, a także inne istotne charakterystyki, takie jak wielkość "szarej strefy", czyli ilość niezdiagnozowanych zakażeń lub ilość zakażeń napływających spoza kraju czy województwa w proporcji do zakażeń wewnętrznych.
Metody wypracowane w aktualnym starciu z koronawirusem pozwolą zrozumieć i opanować przyszłe i bieżące epidemie i pandemie znanych i nieznanych jeszcze wirusów. Aktywna walka z obecną pandemią jest szansą na wypracowanie ścieżki zapobiegawczej na przyszłość.
 Autor: Dr Maciej Sykulski, koordynator ds. naukowych genXone, pionier analiz bioinformatycznych opartych o sekwencjonowanie w technologii nanoporowej. Posiada wieloletnie doświadczenie w dziedzinie bioinformatyki i w prowadzeniu prac badawczo-rozwojowych we współpracy z biotechnologią. Autor wielu międzynarodowych publikacji, brał udział w międzynarodowych projektach naukowych i wdrożeniowych. Jego główne obszary badawcze to: wykrywanie wariantów oraz rearanżacji w genomach ludzkich, analiza danych metagenomicznych mikrobiomu, w tym nowe algorytmy rozpoznawania gatunków i szczepów bakteryjnych, analiza wysokowymiarowa, wydajne i efektywne oprogramowanie bioinformatyczne.
Autor: Dr Maciej Sykulski, koordynator ds. naukowych genXone, pionier analiz bioinformatycznych opartych o sekwencjonowanie w technologii nanoporowej. Posiada wieloletnie doświadczenie w dziedzinie bioinformatyki i w prowadzeniu prac badawczo-rozwojowych we współpracy z biotechnologią. Autor wielu międzynarodowych publikacji, brał udział w międzynarodowych projektach naukowych i wdrożeniowych. Jego główne obszary badawcze to: wykrywanie wariantów oraz rearanżacji w genomach ludzkich, analiza danych metagenomicznych mikrobiomu, w tym nowe algorytmy rozpoznawania gatunków i szczepów bakteryjnych, analiza wysokowymiarowa, wydajne i efektywne oprogramowanie bioinformatyczne.




KOMENTARZE