


Fot. Dr hab. Karolina Mikulska-Rumińska, prof. UMK z Wydziału Fizyki, Astronomii i Informatyki Stosowanej UMK, źródło: Andrzej Romański
Zespół Bartha to choroba skorelowana z chromosomem X. Dotyka chłopców, dziewczynki natomiast mogą być jedynie bezobjawowymi nosicielkami. Rozwija się w pierwszej dekadzie życia, głównie w pierwszym roku od urodzenia. Występuje z częstością ok. 1 na 400 tys. urodzeń. Poza cechami fizycznymi, takimi jak głęboko osadzone oczy, wydatne policzki, wysokie czoło czy odstające uszy, chorzy cierpią na poważne problemy m.in. z sercem, mięśniami szkieletowymi, nie rosną prawidłowo oraz mają problemy neurologiczne, tj. obniżone zdolności wzrokowo-przestrzenne i ograniczone zdolności matematyczne.
Nadzieja na lek
Chociaż pierwszy przypadek został opisany w 1983 r. przez zespół holenderskiego uczonego Petera Bartha, do tej pory nie udało się znaleźć skutecznego leku. Rokowania pacjentów są bardzo złe – większość chorych umiera we wczesnym dzieciństwie. Jest jednak nadzieja – w artykule „Anomalous peroxidase activity of cytochrome c is the primary pathogenic target in Barth syndrome”, który ukazał się 23 listopada br. w „Nature Metabolism” międzynarodowa grupa badaczy i badaczek nie tylko wskazała wiodący mechanizm patogenny, za sprawą którego zespół Bartha się rozwija, ale też wytypowała związek chemiczny, jaki może być wprowadzony do testów klinicznych dla pacjentów z tym schorzeniem.
Awaria w „fabryce energii”
Do tej pory w terapii chorych skupiano się na leczeniu objawów – problemów z sercem, mózgiem i mięśniami. Nieznany był bowiem mechanizm odpowiedzialny za powstanie choroby, naukowcy nie wiedzieli, co dokładnie wpływa na jej rozwój. Testom klinicznym poddane zostały dwa potencjalne leki, jednak bez sukcesów – jeden z nich nie działa wcale, drugi zaś częściowo łagodzi tylko objawy, ale wciąż nie daje szansy na wyleczenie zespołu Bartha. Niemal 40-osobowa grupa badaczy i badaczek postanowiła przyjrzeć się tematowi kompleksowo – począwszy od eksperymentów biochemicznych i biofizycznych, poprzez modelowanie komputerowe, kończąc na testach na zwierzętach oraz badaniach na próbkach z biopsji pacjentów z zespołem Bartha.
Prof. Karolina Mikulska-Rumińska była odpowiedzialna za wyjaśnienie mechanizmów molekularnych leżących u podstaw choroby przy użyciu modelowania komputerowego. Wnioski są przełomowe dla sprawy. – W zespole Bartha mamy do czynienia z mutacją w chromosomie X, a dokładnie – genie TAZ, który koduje tafazynę, czyli białko biorące udział w metabolizmie lipidów, konkretnie kardiolipin. Co należy podkreślić, kardiolipiny są bardzo ważne – występują w mitochondriach, czyli naszych „fabrykach energii” zasilających wiele procesów w naszym organizmie. Jeśli mamy do czynienia z mutacjami w tafazynie, wówczas kardiolipiny nie powstają, a w zamian akumulują się monolizokardilipiny, czyli kardiolipiny bez jednego łańcucha acylowego. Wtedy mamy poważny problem, ponieważ kardiolipiny wpływają na funkcje stu innych białek mitochondrialnych – wyjaśnia polska badaczka. Należy dodać, że kardiolipiny tworzone są z monolizokardiolipin, m.in. przy pomocy tafazyny. W zespole Bartha dochodzi do nagromadzenia monolizokardiolipin i deficytu kardiolipin. Co jednak jest bezpośrednią przyczyną dysfunkcji mitochondriów oraz skutków chorobowych określanych jako zespół Bartha, dotąd naukowcom nie udało się wyjaśnić.
Toksyczna maszyneria
– Udało nam się ustalić, że monolizokardiolipiny, czyli ta uboższa wersja kardiolipin, łączą się z cytochromem c. To bardzo ważne białko, odpowiedzialne m.in. za oddychanie komórkowe, a także biorące udział w programowanej śmierci komórki. Odkryliśmy, że monolizokardiolipina, łącząc się z cytochromem c, zmienia jego funkcje, transformując go w anormalną peroksydazę zdolną do katalizowania peroksydacji mitochondrialnych fosfolipidów PUFA. Innymi słowy, kompleks zaczyna działać jak maszyneria tworząca toksyczne fosfolipidy, które zaczynają wpływać negatywnie właśnie na mięśnie szkieletowe, serce czy mózg. Udowodniliśmy więc, że to ten kompleks – monolizokardiolipiny z cytochromem c – jest podstawą, początkiem zespołu Bartha – tłumaczy prof. Mikulska-Rumińska. Zespół wskazał również konkretny związek chemiczny, który blokuje kompleks cytochromu c z monolizokardiolipiną. Prof. Mikulska-Rumińska pokazała, w jaki sposób ten proces przebiega. Pozwoliło to lepiej zrozumieć podstawy molekularne badanego układu, co jest niezbędne do zaprojektowania efektywnego leku.
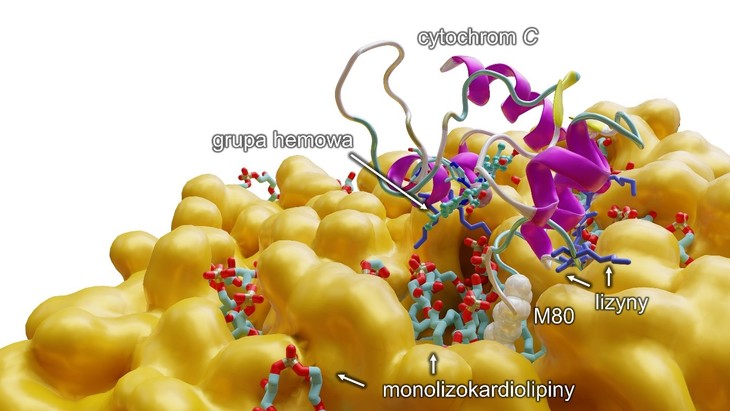
Rys. Modelowanie komputerowe ukazujące na poziomie molekularnym oddziaływanie monolizokardiolipiny z cytochromem c, które skutkuje rozerwaniem wiązania między żelazem z grupy hemowej a metioniną 80 (M80) i przejście cytochromu do pięciokoordynacyjnej formy, aktywując przy tym anormalną funkcjonalność białka, czyli zdolność do peroksydacji wielonienasyconych kwasów tłuszczowych (PUFA). Dodatkowo (na niebiesko) przedstawiono lizyny, aminokwasy o dodatnim ładunku, które pozwalają cytochromowi c przyłączyć się do błony lipidowej (na pomarańczowo). Źródło: UMK
Autorka: Żaneta Kopczyńska, Uniwersytet Mikołaja Kopernika w Toruniu




KOMENTARZE